We discuss an issue with using Langevin thermostat in LAMMPS combined with dynamics that is restarted every single timestep. The system doesn't equilibrate to the correct temperature. A similar issue also arises when using a N-sized loop combined with the langevin thermostat (if the pre keyword isn't set to no). Short molecular dynamics trajectories that are interrupted periodically are typical to hybrid MC-MD calculations and also often occur in other path sampling methods.
First we present the problem and explore how these problems can be fixed.
Simulation Details
We performed NVT simulations of Lennard-Jones particles with $k_B T/\epsilon=T^*=0.96$ and $\rho \sigma^3=\rho^*=0.5$. The simulations are carried out in reduced Lennard-Jones units using the units lj command in LAMMPS. 512 particles are simulated using the lj/cut pairstyle in LAMMPS with the potential shifted to 0 at the cutoff of $2.5\sigma$. The velocities are assigned using the velocity command and a random seed is chosen for the initialization. The timestep chosen for all the simulations is 0.005 and 5 independent blocks are simulated to calculate error bars unless stated otherwise.
Case 1- simulations without restarts
The system is simulated for $10^8$ timesteps with the Nose-Hoover and the Langevin thermostat. Every 1000 timesteps we collect the value of the kinetic energy, potential energy, total energy and the temperature of the system. The histogram of total energies should overlap perfectly irrespective of the choice of the thermostatting procedure. The data from the first $10^6$ timesteps is discarded as equilibration data.
Figure 1- The black points are histograms of total energy calculated using the Nose-Hoover thermostat. The error bars are generated by running 5 independent simulations ($10^8$ timesteps each). The green histogram is generated using the Langevin thermostat. Clearly, the histograms of total energies using the two different thermostats overlap.
The calculated temperature for the Nose-Hoover system is $0.9601 \pm 0.0003$ and for the Langevin system is $0.9616 \pm 0.0005$.
Case 2- simulations with restarts
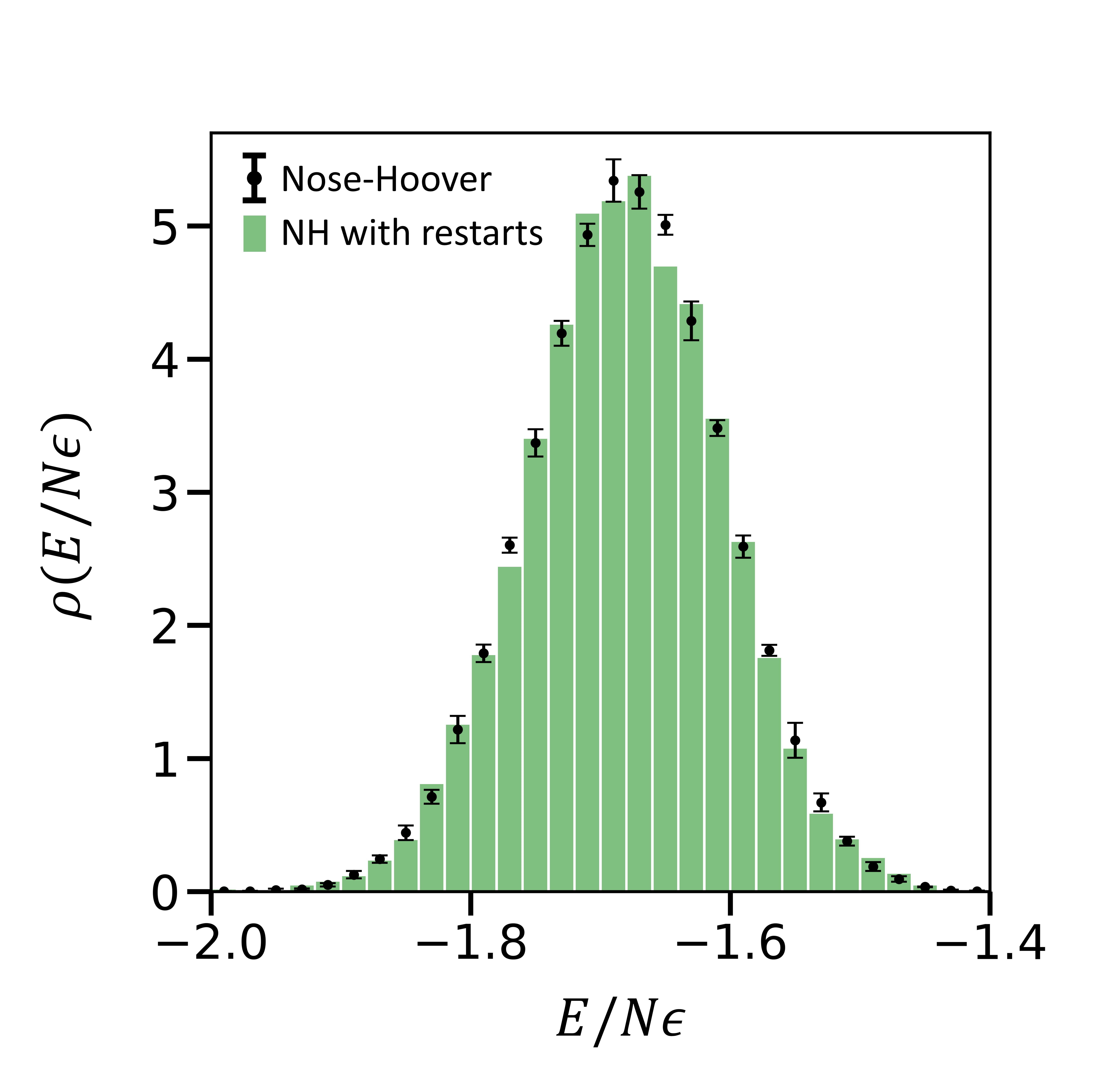
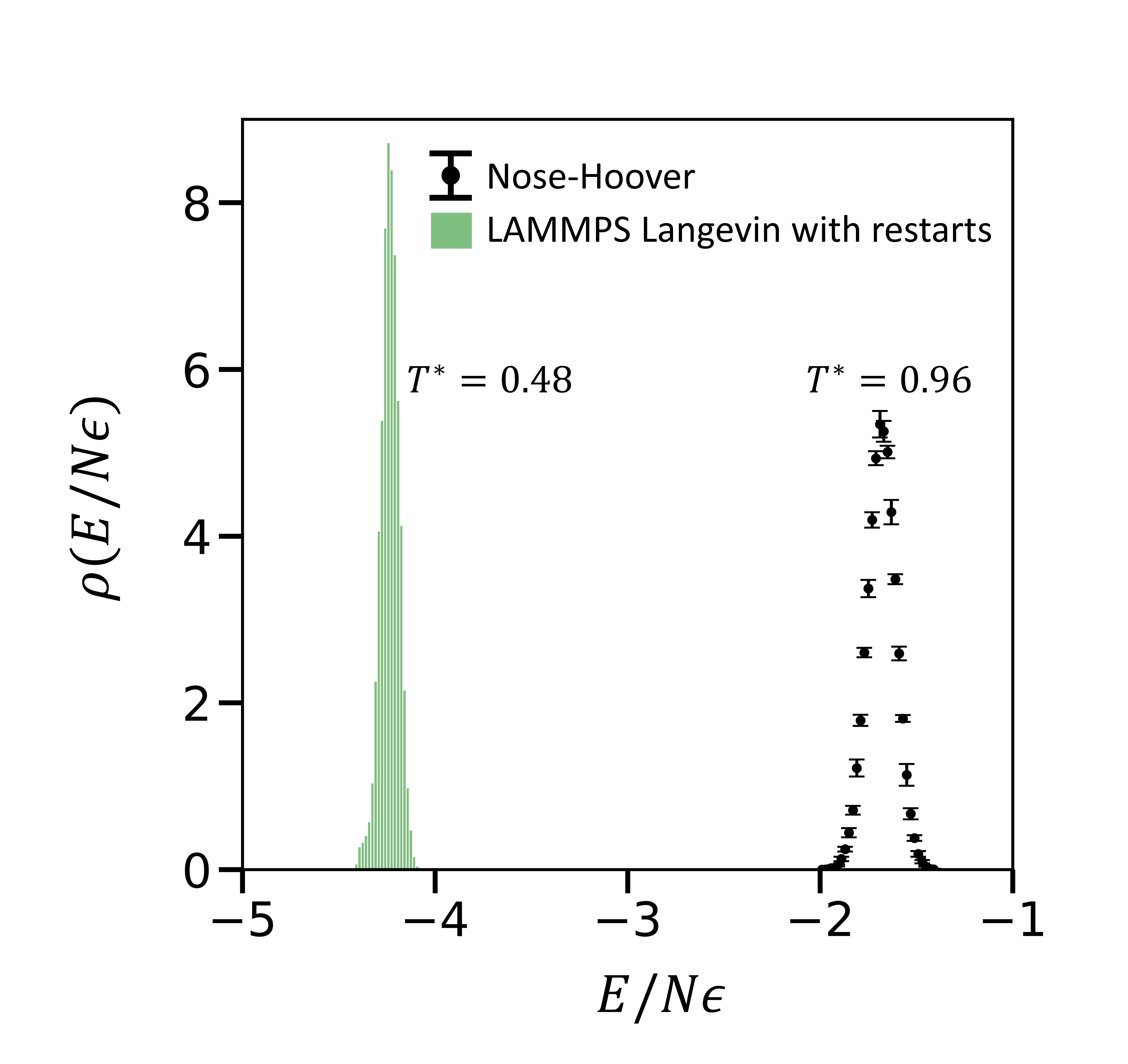
The thermodynamic quantities computed should statistically be the same irrespective of the frequency of writing a restart file and restarting a simulation. In other words, periodic interruption of dynamics shouldn't affect the total energy histograms for a canonical ensemble. We now describe our results for simulations where a restart file is written every timestep using the write_restart command in LAMMPS and restarted using the read_restart command. $10^7$ timesteps are simulated where the thermodynamic quantities are output every 1000 timesteps. The temperature of the Nose-Hoover system with restarts is $0.960 \pm 0.002$ (see figure 2). The ensemble simulated by using Langevin with restarts in LAMMPS does not correspond to the correct canonical ensemble(see figure 3).
Interestingly the temperature of the Langevin system with restarts is exactly half of the setpoint $T^*=0.96$. Thus, the average kinetic energy of the system is lower than the expected value.
Figure 2- The black points are the same as described in figure 1. The green histogram is generated using the Nose-Hoover thermostat with restarts ($10^7$ timesteps). Clearly, the histograms of total energies using the two different procedures overlap.
Figure 3- The black points are the same as described in figure 1 and 2. The green histogram is generated using Langevin with restarts ($10^7$ timesteps).
A related problem also occurs when using the Langevin thermostat in a N-sized loop as follows (pseudocode)
fix 1 all langevin
fix 2 all nve
timestep 0.005
label Step
run 1
print thermodata
jump in.file Step
Interestingly the temperature equilibrates to half the set point as well in this implementation. The two problems above have a simple explaination that is connected to the Langevin algorithm that LAMMPS uses.
The Schneider and Stoll method that LAMMPS uses (with the velocity Verlet algorithm) is
$$
\textbf{v}(t+\delta t/2)=\textbf{v}(t)+\frac{\delta t}{2 m}\Big(\textbf{F}^C(t)-\zeta m \textbf{v}(t-\delta t/2)+\sqrt{\frac{2k_B T \zeta m}{\delta t}}\textbf{g}\Big) \\
\textbf{r}(t+\delta t)=\textbf{r}(t)+\textbf{v}(t+\delta t/2)\ \delta t \\
\textbf{F}(t+\delta t)=\textbf{F}^C(\textbf{r}(t+\delta t))-\zeta m \textbf{v}(t+\delta t/2)+\sqrt{\frac{2k_B T \zeta m}{\delta t}}\textbf{g'} \\
\textbf{v}(t+\delta t)=\textbf{v}(t+\delta t/2)+\frac{\delta t}{2 m} \textbf{F}(t+\delta t)
$$
Here $\textbf{F}^C$ represents the conservative force, $\zeta$ is the friction coefficient, and $\textbf{g}$ and $\textbf{g'}$ are two independent Gaussian normal vectors. The crucial observation to be made in the algorithm is that $\textbf{F}(t+\delta t)$ is used to update the second half of the velocity update in the $n^{th}$ iteration and the first half of the velocity update in the $n+1^{th}$ iteration. This observation is the key to the resolution of the thermostatting problem.
LAMMPS in the restart files saves positions and velocities of the particles in the system. However, for a perfect restart at time $t$ we need access to the Langevin forces applied at $t-\delta t/2$. This is one of the key reasons the fix langevin LAMMPS doc page mentions that exact restarts of the system are not possible using this fix. The thermostatting issue with the N-sized loop is based on a similar reason but has more to do with the subtlety of force updates in LAMMPS. Dr. Plimpton wrote that LAMMPS precomputes the forces during the setup of the run. The Langevin forces are added during the setup before the run starts. The system is propagated and fresh Langevin forces act in the middle of the timestep. If we loop through a run command (as shown in the above pseudocode) the Langevin forces would be recomputed before the run in every iteration. We do not want this as we want to carry forward the total force at the end of the $n^{th}$ iteration to the $n+1^{th}$ iteration. The easy fix to this problem is setting the pre keyword to no. So the modified pseudocode is
fix 1 all langevin
fix 2 all nve
timestep 0.005
run 1
label Step
run 1 pre no
print thermodata
jump in.file Step
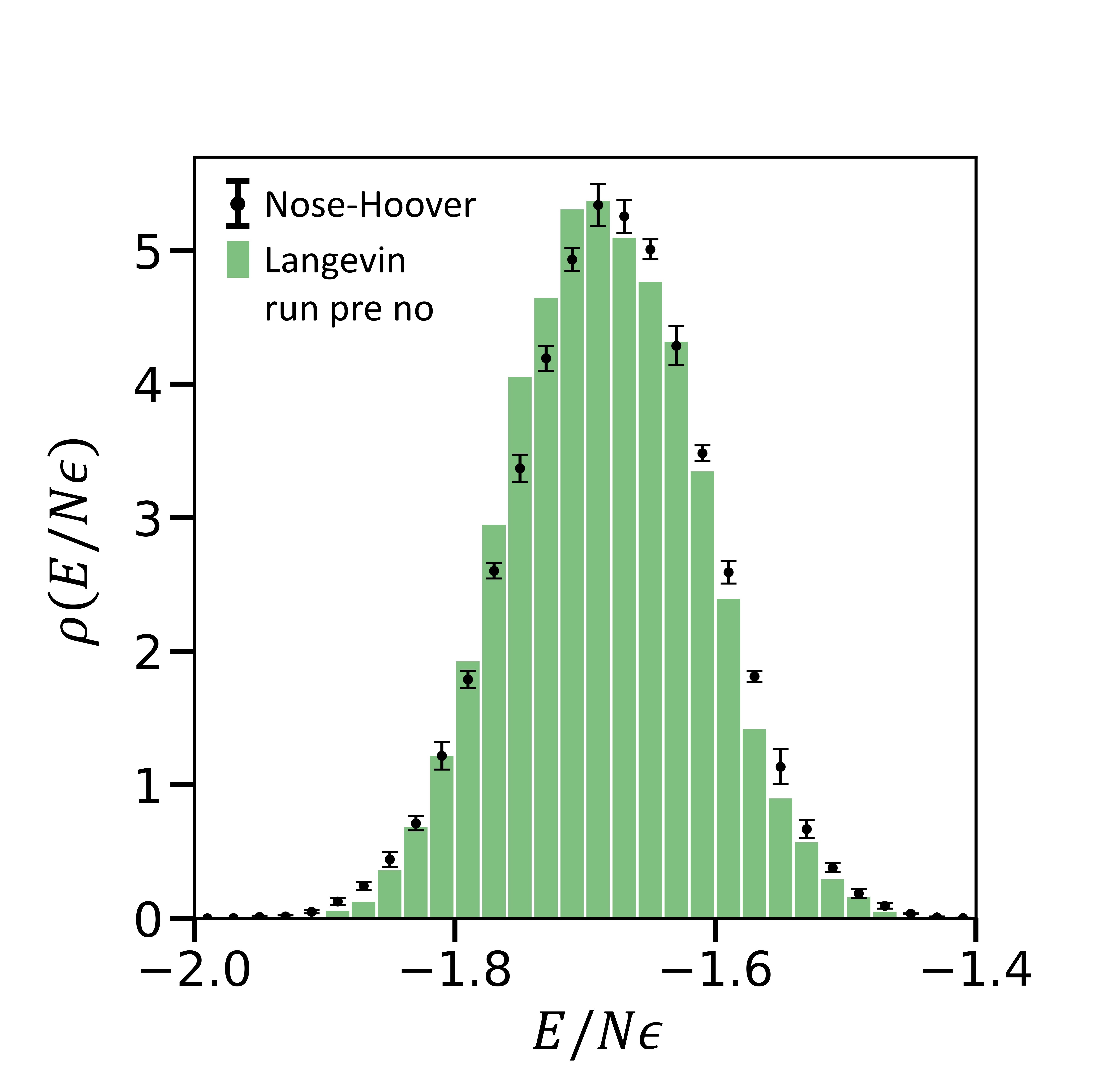
The system equilibrates to the correct temperature ($0.959$) in this case. The figure below compares the total energy histograms.
Figure 4- The black points are the same as described previously (long Nose-Hoover runs). The green histogram is generated using the modified pseudocode above. Clearly, the histograms of total energies using the two different procedures overlap.
Acknowledgements
We thank Steve Plimpton (Sandia National Labs) for prompt correspondence and help in resolution of the problem. Dr. Plimpton gave important suggestions (especially for the case with restart files) that allowed us to implement the calculations within the existing structure of LAMMPS. Additionally, we would also like to thank Prof. Niels Grønbech-Jensen (UC-Davis) for insight into the Schneider and Stoll Langevin algorithm that LAMMPS uses.